
 Gaza: under the spotlight, the Israeli-Palestinian conflict shakes up the Eurovision contest
Gaza: under the spotlight, the Israeli-Palestinian conflict shakes up the Eurovision contest Black soldier killed by a police officer in the United States: the sheriff publishes the video of the arrest
Black soldier killed by a police officer in the United States: the sheriff publishes the video of the arrest In Malmö, the Eurovision party transformed into entrenched camps
In Malmö, the Eurovision party transformed into entrenched camps In Russia, Vladimir Putin stigmatizes “Western elites”
In Russia, Vladimir Putin stigmatizes “Western elites” “Mediterranean diet” or “DASH”, two good tips for eating better
“Mediterranean diet” or “DASH”, two good tips for eating better Fatal case of cholera in Mayotte: the epidemic is “contained”, assures the government
Fatal case of cholera in Mayotte: the epidemic is “contained”, assures the government The presence of blood in the urine, a warning sign of bladder cancer
The presence of blood in the urine, a warning sign of bladder cancer A baby whose mother smoked during pregnancy will age more quickly
A baby whose mother smoked during pregnancy will age more quickly Artificial intelligence lies, cheats and deceives us, and that's a problem, experts warn
Artificial intelligence lies, cheats and deceives us, and that's a problem, experts warn Google Cloud mistakenly deletes UniSuper fund account and deprives 600,000 Australians of their superannuation
Google Cloud mistakenly deletes UniSuper fund account and deprives 600,000 Australians of their superannuation IBM, Amazon, Hager... These record investments expected at the Choose France summit
IBM, Amazon, Hager... These record investments expected at the Choose France summit Boeing's black streak: a second Air France flight diverted in three days for “a smell of heat”
Boeing's black streak: a second Air France flight diverted in three days for “a smell of heat” Paola Locatelli: “Influence is comfortable but cinema gives me adrenaline”
Paola Locatelli: “Influence is comfortable but cinema gives me adrenaline” Swifties attack hotels in Lyon
Swifties attack hotels in Lyon Radical Optimism by Dua Lipa, a tangy album in half-tone
Radical Optimism by Dua Lipa, a tangy album in half-tone Peter Weir rewarded for his career with an honorary Golden Lion at the Venice Film Festival in September
Peter Weir rewarded for his career with an honorary Golden Lion at the Venice Film Festival in September Omoda 7, another Chinese car that could be manufactured in Spain
Omoda 7, another Chinese car that could be manufactured in Spain BYD chooses CA Auto Bank as financial partner in Spain
BYD chooses CA Auto Bank as financial partner in Spain Tesla and Baidu sign key agreement to boost development of autonomous driving
Tesla and Baidu sign key agreement to boost development of autonomous driving Skoda Kodiaq 2024: a 'beast' plug-in hybrid SUV
Skoda Kodiaq 2024: a 'beast' plug-in hybrid SUV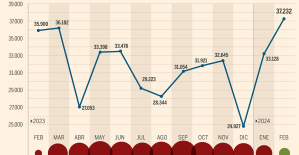 The home mortgage firm rises 3.8% in February and the average interest moderates to 3.33%
The home mortgage firm rises 3.8% in February and the average interest moderates to 3.33%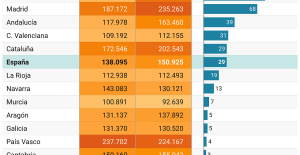 This is how housing prices have changed in Spain in the last decade
This is how housing prices have changed in Spain in the last decade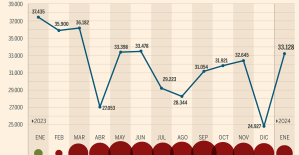 The home mortgage firm drops 10% in January and interest soars to 3.46%
The home mortgage firm drops 10% in January and interest soars to 3.46% The jewel of the Rocío de Nagüeles urbanization: a dream villa in Marbella
The jewel of the Rocío de Nagüeles urbanization: a dream villa in Marbella Diving into the secrets of the National Assembly
Diving into the secrets of the National Assembly Institutions: senators want to restore the accumulation of mandates and put an end to the automatic presence of ex-presidents on the Constitutional Council
Institutions: senators want to restore the accumulation of mandates and put an end to the automatic presence of ex-presidents on the Constitutional Council Europeans: David Lisnard expresses his “essential and vital” support for François-Xavier Bellamy
Europeans: David Lisnard expresses his “essential and vital” support for François-Xavier Bellamy Facing Jordan Bardella, the popularity match turns to Gabriel Attal’s advantage
Facing Jordan Bardella, the popularity match turns to Gabriel Attal’s advantage These French cities that will boycott the World Cup in Qatar
These French cities that will boycott the World Cup in Qatar Sailing: the Lecointre/Mion and Picon/Steyaert duos European medalists
Sailing: the Lecointre/Mion and Picon/Steyaert duos European medalists Liga: Atlético Madrid beats Celta Vigo and consolidates its fourth place
Liga: Atlético Madrid beats Celta Vigo and consolidates its fourth place Foot (F): Lyon crushes Reims and joins PSG in the championship final
Foot (F): Lyon crushes Reims and joins PSG in the championship final Serie A: when Italian clubs flock jerseys with the first names of players' mothers to celebrate Mother's Day
Serie A: when Italian clubs flock jerseys with the first names of players' mothers to celebrate Mother's Day
One person in 5,000 - and the result, perhaps, of major arterial problems. Marfan syndrome affects around 13,000 people in France. It is a disease resulting from a mutation in the FBN1 gene located on chromosome 15. It is therefore transmissible to descendants via a mode of transmission called “autosomal dominant”: autosomal, because the mutation is located on a autosomal chromosome, that is to say non-sexual (X or Y). Dominant, because a mutation in one of the two copies of the gene is enough to trigger the disease.
The FBN1 gene encodes a protein called “fibrillin 1.” The latter allows the organization of elastin fibers constituting the extracellular matrix of connective tissue, whose role is to ensure organ support. Connective tissue is present everywhere in the body, and this explains why this disease causes a wide variety of manifestations and can affect the heart, eyes, skeleton or skin.
The clinical signs are not the same in all people, even within the same family. The main danger of Marfan syndrome is that it can damage the aorta, the body's main artery which carries blood from the heart to all parts of the body. Due to the greater elasticity of the tissues in patients, the artery will tend to dilate, which can ultimately cause aortic dissection: a partial tear in the wall of the aorta.
Professor Guillaume Jondeau is a cardiologist at Bichat-Claude-Bernard hospital and coordinator responsible for the Reference Center for Marfan syndrome and related diseases. He gives an optimistic assessment: “Since the 1970s, we have made enormous progress, the leading cause of death is aortic dissection, and the leading cause of dissection is the absence of diagnosis. Today, we detect better, we monitor better, we operate on time. We give beta blockers to patients, we avoid brutal and violent efforts which increase tension and promote dissection. Thanks to this, we have gained a lot of life expectancy. Indeed, in the 1970s, there were still 50% deaths at the age of 40, whereas today patients live as long as the general population.”
There is no treatment for the disease yet, but progress is real. It is possible to properly measure the risk using echocardiography and CT scans. The primary goal is to slow the dilation of the aorta by maintaining normal blood pressure using beta blockers which slow the heart. If the dilation is too severe, surgery may be performed. The principle of all interventions is to replace the initial part of the aorta, which is the most fragile area. This type of operation is carried out approximately once every two weeks at the Marfan Syndrome Reference Center, and Professor Jondeau is pleased with a 100% success rate in the last 35 surgeries.
Research continues to progress on diseases related to Marfan syndrome, and today it is no longer a single gene that is studied, but a panel of 38 genes responsible for genetic diseases of the connective tissue. This more detailed knowledge could help provide better patient care. This is why Professor Jondeau calls on all people with an aortic aneurysm or signs suggestive of Marfan to consult a reference or competence center.


















